Studying the History of Character
Evolution
With a phylogenetic tree and a distribution
of character states in the observed (terminal) taxa, Mesquite
can attempt to reconstruct the character states at ancestral nodes.
Two separate issues to consider are the method by which the reconstruction
is done, and how its results are displayed to the user. Mesquite
currently can use either parsimony or likelihood to reconstruct
ancestral states, and has several display methods, including "Trace
Character History" which paints the branches of the tree
to show the reconstruction.
We recommend highly that you examine
the example files provided in the folder "Ancestral
State Examples". The minimal configuration to use with
these examples is "Ancestral States" (indicate
this configuration under
File>Activate/Deactivate Packages>Choose
Configuration), but you can also leave Mesquite in
its default All Installed Modules mode.
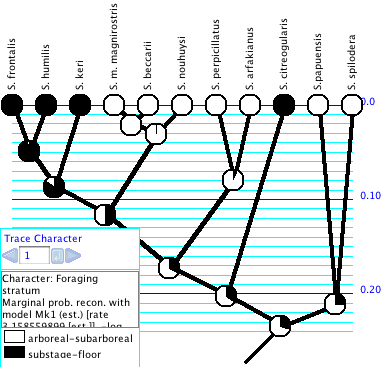
Trace Character History
The Trace Character History facility
graphically represents a history of character evolution on
the
tree. It is available under the Analysis menu of a tree window
(e.g., the basic Tree Window, Dependent Tree Window, Mirror
Tree
Window, Multitree Window). If you select this you will probably
be asked for a source of characters (e.g., stored characters)
and a reconstruction method (e.g., parsimony). The tree will
be painted to show ancestral states, and a trace legend will
appear.
The trace legend contains an important text area that gives details
of the current ancestral state tracing. You can also see details
of the reconstruction by switching the window to Text mode
using the tabs at its top.
The Trace menu gives menu items to
control the character history and its display. Some important
ones are:
Character history source:
The character history displayed can be reconstructed using observed
states in terminal taxa ("Reconstruct Ancestral States")
or can be a simulated history ("Simulate Ancestral States").
The reconstructed states need not be based on actual data, but
could be based on simulated data. "Simulate Ancestral States"
shows the "actual" history of character evolution branch
by branch as it occured in the simulation,
not as it was reconstructed, and thus may show ancestral states
that would be unreconstructable, obliterated by subsequent changes.
Next, Previous, Choose Character
History: Usually, this will allow you to choose which
character to view. You can also scroll through characters using
the blue arrows in the trace legend.
Trace Display Mode:
With Trace>Trace Display Mode>Shade States
and Trace>Trace Display Mode>Label States
you choose whether to see the branches painted, or have the states
indicated in labels. If painted, you can also ask that states
be indicated by labels by choosing Trace>Label
States.
You can also see details of the reconstruction
at a node by holding the cursor over the branch. A description
of the reconstructed states will appear at the bottom part
of
the Trace Character Legend. Another method is to use the Text
view of the window (touch on the Text tab at the top of the
tree
window) and scroll down — a text version of the trace should
appear.
Trace All Characters
Trace All Characters summarizes ancestral state reconstructions
of many characters simultaneously. To request it, choose Choose (Tree
Window)Analysis>Trace All Characters. A text window
like that shown below will appear, listing the ancestral states
reconstructed at each node for each character. Node numbers show
up in red on the tree. (Alternatively, spots showing
node numbers in the figure below can be turned on in the Tree
Window's
Drawing
menu by selecting Show Node Numbers.)
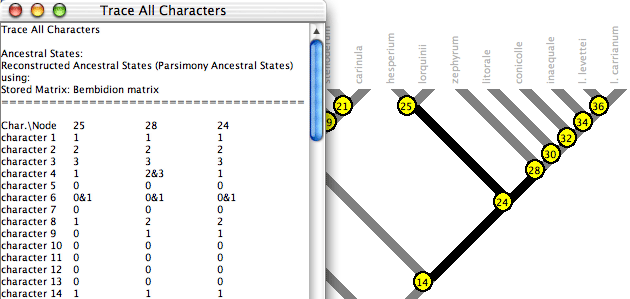
By default only the selected nodes are listed.
(Nodes can be selected using tools in the Tree Window.) You can
request to show all nodes by turning off Show Selected Nodes Only in the Trace_All menu. By default
all characters are listed; this can be changed using the Show Selected Characters Only menu item.
The ancestral state reconstruction can be controlled
in the Trace_All menu of the tree window.
The table is either listed by characters or by nodes; you can switch from one to the other using the Rows are Characters
menu item
Columns in the table in the text window may not
appear perfectly aligned, but it is presented as a tab-delimited
table, so you should be able to copy the text and paste it in
to a text file to read in to your favorite spreadsheet program.
Trace Character Over Trees
The Trace Character Over Trees facility summarizes ancestral
state
reconstructions over a series of trees. This is useful to understand
how ancestral state reconstructions vary over a series of
trees,
for instance if there is uncertainty in the tree. It works for
categorical characters only. Also, Trace Character
Over Trees does NOT calculate a consensus tree for
you. As with all other analyses in the Tree Window, it works
with the tree
that is given to it by the Tree Window. If you want to make your
summary on a consensus tree, then you need to put the consensus
tree
into
the Tree Window
first and then request Trace Character Over Trees.
Choose (Tree Window)Analysis>Trace
Character Over Trees. This examines a series of trees,
and for each examines a character's ancestral states on that tree.
For each node in the tree in the tree window, it attempts to summarize
what ancestral states are reconstructed for that same clade in
the series of trees (as long as the same clade exists in the other
trees). For example, imagine the tree in the tree window includes
the clade Tetrapoda. Each of the series of trees is examined,
and if that tree includes the clade Tetrapoda, then its reconstructed
ancestral states are examined. If the tree doesn't include Tetrapoda,
then it is ignored for the sake of summarizing the tetrapod ancestral
states. The tree in the tree window is then decorated to summarize
what ancestral states are reconstructed for each of the clades.
Here is an example of Trace Character Over Trees in action:
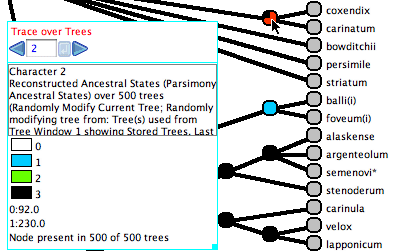
The cursor is over a node, and thus the legend shows a summary.
The node (i.e., the clade it represents) is present in all 500
of the trees examined. In 92 of those trees, state 0 is reconstructed
at the node. In 230 trees, state 1 is reconstruced.
These two numbers do not add to 500 because some trees show an
ambiguous (equivocal) ancestral state reconstruction at the
node, and the option to count only the Uniquely Best States is
selected
in the Count submenu of the Trace_Over_Trees menu. With this
option a tree is counted as having a state at a node only if
the state is the only optimal state. What is considered optimal
depends on the reconstruction method. With parsimony,
states are considered equally optimal if they are equally parsimonious.
With likelihood, the Decision Threshold is used to decide whether
states are good enough to be considered within the optimal set.
The alternative to "Uniquely Best State" is "Equivocal". With
the Equivocal option for counting, a tree is counted as having a state at a node if
the state is within the optimal set, whether or not there are other states
within the optimal set. When the Equivocal option is used, the sum of tree counts for the states
at a node
can more than the total number of trees with the clade, for a tree can get counted multiply at a node, under each state
in an equivocal assignment.
An important option is what trees to
examine. If the tree in the tree window is a consensus tree, then
the trees examined might be the original set of most parsimonious
trees that built the consensus. Trace Character Over Trees could
then show how the ancestral state reconstruction varies among
the most parsimonious trees. The trees examined might also be
derived from a Bayesian analysis, and the ancestral states obtained
by likelihood, to do an analysis in Lutzoni's style. The trees
might be random resolutions of an unresolved tree, or trees with
random noise added to branch lengths, and so on. This would allow
you to see how the results would vary if the tree changed. See
the submenu Trace_Over_Trees>Tree Source
for options.
Parsimony Reconstruction Methods
Parsimony reconstruction methods find
the ancestral states that minimize the number of steps of character
change given the tree and observed character distribution. They
can use different assumptions (models of evolution). For categorical
characters, the unordered states assumption is
that one step is counted for any change. The ordered
states assumption is that the number of steps from state i to
state j is |i-j|. Thus, the number of steps from state 2 to state
5 is 3 steps. A stepmatrix explicitly specifies
the number of steps from state to state by a matrix. Mesquite
does not yet do parsimony calculations for irreversible,
Dollo and character state tree
assumptions, although these models are listed in menus and you
can assign them to particular characters. For continuous
characters, the linear cost assumption is that
the cost of a change from state x to state y is |x-y|. The squared
change assumption is that the cost of a change from state x to
state y is (x-y) squared.
Mesquite's parsimony calculations attempt
to match MacClade's. Some differences remain in special cases
of polymorphic terminal taxa with stepmatrices. Mesquite allows
hard polytomies in the tree when stepmatrices are used.
Assigning a parsimony model:
The parsimony model used for a character's calculations is the
model assigned to it, if the character is one stored in in a matrix
in a file. A parsimony model can be assigned in the List of Characters
window. Select the row(s) corresponding to the desired character(s),
and then touch on the column heading "Parsimony Model".
A drop-down menu contains a submenu that allows you to select
the models to apply. You can also change the parsimony model assigned
to the character being traced in Trace Character History using
the Parsimony Model submenu of the Trace menu. (Recall that Mesquite
cannot yet do calculations with irreversible and Dollo models.)
If the characters used in parsimony reconstruction are not stored
in a matrix but rather come directly from another source of
characters
such as simulations, a single parsimony model can be chosen to
be applied to all of the characters coming from this source.
Thus,
for instance, when using Trace Character History, the Parsimony
Model submenu of the Trace menu can be used to assign the model
to be used.
Creating and editing stepmatrices:
To create a stepmatrix, select Characters>New
Character Model>Stepmatrix. A window will appear
in which you can edit the cost of i to j transitions. The
number
of states allowed is initially 10 (0 through 9), but you can
change the number of states under (Edit
Stepmatrix)>Step_matrix>Set
maximum state. The maximum number of states for a
categorical character is 56; the maximum state value is therefore
55. This stepmatrix editor does not do triangle inequality
checking (see discussion in manual of MacClade, which does check
the triangle inequality).
The parsimony calculations are used
also for Treelength and Character Steps.
Likelihood Reconstruction Methods
Likelhood reconstruction methods find
the ancestral states that maximize the probability the observed
states would evolve under a stochastic model of evolution (Schluter
et al., 1997; Pagel, 1999). The likelihood reconstruction finds,
for each node, the state assignment that maximizes the probability
of arriving at the observed states in the terminal taxa, given
the model of evolution, and allowing the states at all other nodes
to vary. (In fact, this considers all possible assignments to
the other ancestral states.) This is equivalent to the marginal
reconstruction of Swofford's PAUP*, or the Fossil Likelihood reconstruction
of Pagel's Discrete.
You can use likelihood for the reconstruction
by selecting "Likelihood Ancestral States" when first requesting
Trace Character History, or from the Method submenu of the Trace
menu after Trace Character History is already active. When using
Likelihood Ancestral States in Trace Character History, it is
recommended that you use a Tree Form for the drawing that uses
spots at the nodes (for example, (Tree Window)Drawing>Tree
Form>Balls & Sticks). These spots at the nodes
will indicate relative likelihoods with pie diagrams as in Schluter
et al. 1997.
At present only categorical
characters are supported by the likelihood calculations. Models
for DNA and protein evolution are not yet available for use
by likelihood. Two models of evolution are currently supported,
the Mk1 model
and the AsymmMk
model.
- Mk1 model
("Markov
k-state 1 parameter model") is a k-state generalization
of the Jukes-Cantor model, and corresponds to Lewis's (2001)
Mk model.
The single parameter is the rate of change. Any particular
change (from state 0 to 1 or state 3 to 2, for example)
is equally
probable. Mesquite's rate of change parameter is equivalent
to the q values of Pagel's Multistate program when the
q's are
constrained to be equal. Thus for a character with three states
0, 1 and 2, the Mk1 model would have an instantaneous rate
matrix
of the following form:
| To |
0 |
1 |
2 |
| From 0 |
- |
q |
q |
| 1 |
q |
- |
q |
| 2 |
q |
q |
- |
- AsymmMk model ("Asymmetrical
Markov k-state 2 parameter model") has two parameters:
one for the rate of increase in state (from 0 to 1, 1 to 2,
etc.; the "forward" rate) and one for the rate of
decrease in state (from 2 to 1, 1 to 0, etc.; the "backward"
rate). For more than two states, this seems a biologically unlikely
model, but for two states is provides a simple model that allows
a bias in gains versus loses. Mesquite supports two alternative
ways to describe the model. The two parameters can be forward
rate and backward rate, or overall rate and bias of gains versus
losses. Thus, if forward and backward rates are both 0.5, then
this can alternatively be described as a rate of 0.5 and a bias
of 1.0 (i.e., unbiased). The conversion between the two representations
is done by the following formulas: forward rate = overall rate
* square root(bias); backward rate = overall rate / square root(bias).
This conversion means that forward rate / backward rate = bias.
Thus an AsymmMk model would have an instantaneous rate matrix
of the following form for a 3-state character:
| To |
0 |
1 |
2 |
| From 0 |
- |
f |
f |
| 1 |
b |
- |
f |
| 2 |
b |
b |
- |
This may be an unrealistic model for 3 or more states (why should
all increases in state number have the same rate and decreases
a different rate?), and so we expect it will be used mostly
for two-state characters.
Many programs bundle the rate of evolution
into the branch lengths of the tree itself. Thus, to change the
rate of evolution, the tree needs to be stretched or shrunk; there
is no separate rate parameter that belongs to the stochastic model
of evolution. This works well as long as the branch lengths are
understood in the same way by the model and the tree, i.e., the
tree's time units (calibration of time scale) are the same as
that of the model. However, in Mesquite different calculations
might make different assumptions about the time scale: coalescence
calculations might need the tree's branches measured in generations,
while a Jukes Cantor model might assume they are in expected nucleotide
substitutions. Thus, many stochastic models in Mesquite have an
extra parameter compared to other programs: the scaling of the
model to the tree. For this reason Mk1 has a rate parameter to
scale the rate against the tree.
If parameters of a model are unspecified,
Mesquite currently estimates them based on the data. Note:
Mesquite currently
estimates parameters on each character separately, not on the
entire data matrix. In addition Mesquite's likelihood calculations
do NOT estimate branch lengths. They use pre-existing branch
lengths
(if a branch length is unassigned, it is treated as 1.0).
Mesquite cannot do likelihood calculations in trees with soft
polytomies, or if some taxa have missing data, polymorphisms,
uncertain states, or gaps in the character. The calculations
also require that the states of a character are contiguous from zero; i.e.,
the character cannot have only states 0,1 and 3.
Other programs that reconstruct ancestral
states using likelihood are Pagel's Discrete and Swofford's PAUP*.
Making, editing and applying
probability models: To use the likelihood calculations,
stochastic (probabilistic) models of evolution must be defined.
Two models are predefined: a general Mk1 model and a general AsymmMk
model. Both of these have their parameters unspecified.
You can also create your own models
and specify their parameters by selecting Characters>New
Character Model>Markov k-state 1-parameter model
(to make an Mk1 model) or Characters>New
Character Model>Asymmetrical 2-param. Markov-k model
(to make an AsymmMk model). In either case a window will appear
in which you can specify the parameters. The Mk1 model allows
you to change the rate. Also, you can change the maximum state
allowed using a menu item in the Mk1_model menu (e.g.,
to restrict
it to binary
characters,
choose 1 as the maximum state). The AsymmMk model allows you
to
change the forward and backward rates, and the maximum state
allowed (using a menu item in the AsymmMk_Model menu). You can
also choose to express the two parameters in the AsymmMk model
as
a
rate (which controls both forward and backward rates) and a bias
(which controls the ratio of forward to backward rates). A bias
of greater than 1 means forward changes are more probable; a
bias
of less than 1 means that backward changes are more probable.
After creating a model, you can edit
it by selecting it under Characters>Edit
Character Model. You can rename or delete a model by
going to the List of Character Models window available under Characters.
Once models are defined they can be
applied and used. When setting up a likelihood calculation,
if
you indicate to use "Stored Probability Model",
the calculation will use the selected model for all characters.
Alternatively, if the characters used are stored in a matrix
(instead of generated temporarily such as by simulations), then
each character
can be assigned a model in advance of the calculation. This can
be done by going to the List of Characters window, selecting
the
row(s) corresponding to the desired character(s), and then touching
on the column heading "Probability Model". A drop-down
menu contains a submenu that allows you to select the models
to
apply. These models will remain assigned to the characters if
you save and reopen the file. You can also change the current
probability model applied to a character by selecting a module
in the Probability Model submenu of the Trace menu. Once models
are assigned to the characters, then the these are treated as
the "Current" models applied to the characters. To
indicate that the likelihood calculations use these assigned
models, indicate
"Current Probability Model" when asked for the source
of models.
Optimization Settings: Models used for likelihood
calculations may have adjustable settings for the optimization
routines used to estimate parameter values. These settings can
be changed under Characters>Model Settings; once changed
the settings are universal, applying to all calculations with
that
category of model. They are stored in the preferences directory
and used again the next time you start Mesquite. The Mk1
Model has one setting: The coarseness of the intervals surveyed
in optimizing the rate parameter. Wider intervals
may result in
finding the optimum more quickly when rates are high, but may
make less accurate when rates are low. Mesquite by default tries
the optimization twice, first with width 1.0 then again with width 10.0,
and then chooses the best results. You can request that Mesquite try a single
fixed width; suggested values are 1.0
to 20.0. To request Mesquite that use its default strategy, enter a width of 0. The AsymmMk model
has one setting, concerning the values of parameters used as
starting
points in the search for optimal parameter values. This setting
is used when both parameters of the model are unspecified and
need to be estimated. There are three options: (1) The rate is
first estimated using the Mk1 model, and then the optimization
routine is given that rate plus a bias of 1.0 as starting values.
(2) The forward and backward rates of 1.0 and 1.0 are used as
starting values, and then 1.0 and 0.1, and then 0.1 and 1.0.
Of these three attempts, the parameter values from the attempt
yielding
the highest likelihood is chosen. (3) Options 1 and 2 are combined,
resulting in four attempts to estimate parameters, first using
the Mk1 rate then the three alternative forward and backward
rates as starting values. Of the four attempts, the parameter
values
from the attempt yielding the highest likelihood is chosen. The
default option is (1). Because the AsymmMk sometimes uses the
Mk1 model, changing the setting of the Mk1 model may affect results
from the AsymmMk model.
Reporting of results: Likelihood ancestral
state reconstructions can be reported in various ways. A first
issue is whether only the best estimates are shown at a node
or instead the support for each state is shown
at a node, regardless of how strong or weak. This is controlled
in Trace Character History by the Display Proportional to Weights
menu item. If it is selected, then support for each state is
shown; otherwise,
only the states judged best are shown. The judgment of what are
the best states is made according to a decision threshold T,
such that if the log likelihoods of two states differ by T or
more, the one with lower
likelihood (higher negative log likelihood) is rejected. This
is set using the Likelihood Decision Threshold menu item.
What states are judged best can be viewed using Trace Character History
in several ways: (1) When the cursor is held over a branch and
the list of states appears at the bottom of the Trace Legend,
the states judged best according to the threshold are marked
with an asterisk; (2) In the Text view of the
window, the list of reconstructions shows
an asterisk by each state judged best at the mode; and (3) When
Display Proportional to Weights is turned off, only the best
states are shaded on the branches. The threshold and best states
are also used in
Trace Character Over Trees.
Another issue is whether the likelihoods of alternative states are reported as is (Raw Likelihoods) or not. The other two
options are to report proportional likelihoods (likelihoods of states are scaled so that they add up to 1, and thus for each state
is shown its proportion of total likelihood) or negative log likelihoods. The former are convenient for interpretation and visualization;
the latter may be more easily used in statistical tests.
Comparison and Interaction with other
Programs
NEXUS-based programs:
Mesquite currently saves the models of evolution for likelihood
in the private MESQUITECHARMODELS block in NEXUS files. A private
block is used because there is as yet no standard for designating
such models. Thus, PAUP*, MrBayes and other programs doing likelihood
calculations will not be able to access these character models.
Comparison with MacClade:
Users familiar with MacClade (macclade.org)
will notice some of its features missing from Mesquite, and vice
versa. MacClade is restricted to parsimony reconstructions, but
has the following features that Mesquite currently lacks. MacClade's
Trace Character facility has the ability to fix states at a node
(the paintbrush tool) and to show individual MPR's (MPRs mode,
formerly Equivocal Cycling). MacClade's Trace All Changes mode
and Changes & Stasis chart summarize reconstructed changes
in all characters. Parsimony models include Dollo, irreversible
and character state trees. Mesquite, on the other hand, includes
likelihood reconstructions, reconstructions for continuous characters
better integrated with Trace Character, branch-length sensitive
calculations and other features such as Trace Over Trees.
Pagel's
Discrete and Multistate programs:
Pagel's Discrete and Multistate programs also do likelihood reconstructions
of ancestral states. Discrete's Fossil Likelihoods with the Global
option corresponds to Mesquite's Likelihood Ancestral States.
To obtain reconstructions in Multistate equivalent to Mesquite's,
define the restricted model equivalent to Mk1 or AsymmMk, using
a series of "restrict" commands to set the q parameters
equal as appropriate. Use the "test" command to estimate
the q rates. Then, restrict the rates to those estimated. Next,
use the "fossil" command to fix the node of interest
to each of the states in turn, after each using the "test"
command to ask Multistate to evaluate the likelihood. These likelihoods
are the global likelihoods for the states at the node.
Discrete and Multistate have several
features not available currently in Mesquite, including the Local
option for parameter estimation, more complete reporting of statistics
for the reconstructions, and calculations to test correlation
among characters using likelihood ratio tests.
Mesquite can import and export data
files for Discrete and Multistate (ppy files). To import, select
the file with Mesquite and choose Pagel format in the import dialog
box. To export, select File>Export....
Acknowledgments
David Swofford assisted by providing
code in C, translated by us to Java, for the optimization routines
used in the likelihood reconstruction.
References
Lewis, P.O. 2001. A likelihood approach to estimating phylogeny
from discrete morphological character data. Systematic Biology
50:913-925.
Lutzoni F, M. Pagel & V. Reeb. 2002. Major fungal lineages
are derived from lichen symbiotic ancestors. Nature 411: 937-940.
Maddison, D.R. and W.P. Maddison. 2000. MacClade version 4:
Analysis of phylogeny and character evolution. Sinauer Associates,
Sunderland Massachusetts.
Pagel, M. 1999. The maximum likelihood approach to reconstructing
ancestral character states of discrete characters on phylogenies.
Systematic Biology. 48: 612-622.
Pagel, M. 2000. Discrete, version 4.0. A computer program distributed
by the author.
Pagel, M. 2002. Multistate, version 0.6. A computer program distributed
by the author.
Schluter D, T. Price, A.O. Mooers, D. Ludwig. 1997. Likelihood
of ancestor states in adaptive radiation. Evolution. 51: 1699-1711.
Swofford, D.L. 2002. PAUP*. Phylogenetic Analysis Using Parsimony
(*and Other Methods), Version 4.0. Sinauer Associates, Sunderland,
Massachusetts.